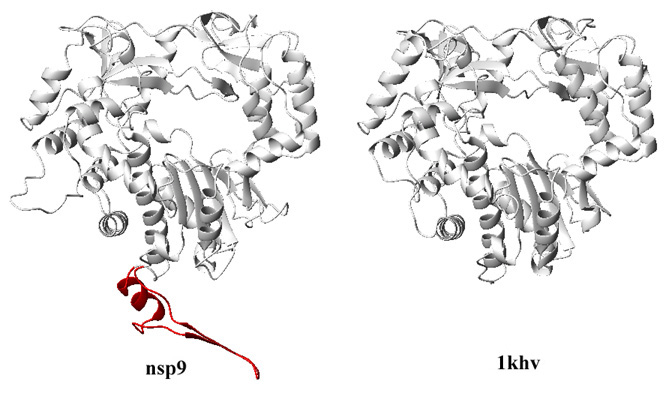
Example 2. SARS nsp9 (polymerase)
I. Using Fold Recognition to find the
appropriate templatesII. Model Building - 1st trial
III. Model evaluation
Primary sequence of nsp9:
>nsp9
SADASTFLNRVCGVSAARLTPCGTGTSTDVVYRAFDIYNEKVAGFAKFLK
TNCCRFQEKDEEGNLLDSYFVVKRHTMSNYQHEETIYNLVKDCPAVAVHD
FFKFRVDGDMVPHISRQRLTKYTMADLVYALRHFDEGNCDTLKEILVTYN
CCDDDYFNKKDWYDFVENPDILRVYANLGERVRQSLLKTVQFCDAMRDAG
IVGVLTLDNQDLNGNWYDFGDFVQVAPGCGVPIVDSYYSLLMPILTLTRA
LAAESHMDADLAKPLIKWDLLKYDFTEERLCLFDRYFKYWDQTYHPNCIN
CLDDRCILHCANFNVLFSTVFPPTSFGPLVRKIFVDGVPFVVSTGYHFRE
LGVVHNQDVNLHSSRLSFKELLVYAADPAMHAASGNLLLDKRTTCFSVAA
LTNNVAFQTVKPGNFNKDFYDFAVSKGFFKEGSSVELKHFFFAQDGNAAI
SDYDYYRYNLPTMCDIRQLLFVVEVVDKYFDCYDGGCINANQVIVNNLDK
SAGFPFNKWGKARLYYDSMSYEDQDALFAYTKRNVIPTITQMNLKYAISA
KNRARTVAGVSICSTMTNRQFHQKLLKSIAATRGATVVIGTSKFYGGWHN
MLKTVYSDVETPHLMGWDYPKCDRAMPNMLRIMASLVLARKHNTCCNLSH
RFYRLANECAQVLSEMVMCGGSLYVKPGGTSSGDATTAYANSVFNICQAV
TANVNALLSTDGNKIADKYVRNLQHRLYECLYRNRDVDHEFVDEFYAYLR
KHFSMMILSDDAVVCYNSNYAAQGLVASIKNFKAVLYYQNNVFMSEAKCW
TETDLTKGPHEFCSQHTMLVKQGDDYVYLPYPDPSRILGAGCFVDDIVKT
DGTLMIERFVSLAIDAYPLTKHPNQEYADVFHLYLQYIRKLHDELTGHML
DMYSVMLTNDNTSRYWEPEFYEAMYTPHTVLQ
Is it possible to use homology modelling to predict the structure of nsp9? Why not?
I. Using Fold
Recognition to find the appropriate templates
1. Go to bioinfo.pl meta-server.Enter your email address, target name and the primary sequence of the target protein.
2. Please do not press the submit button now because we do not want to abuse the server. Instead, I have submitted the job earlier. Click here to view the result.
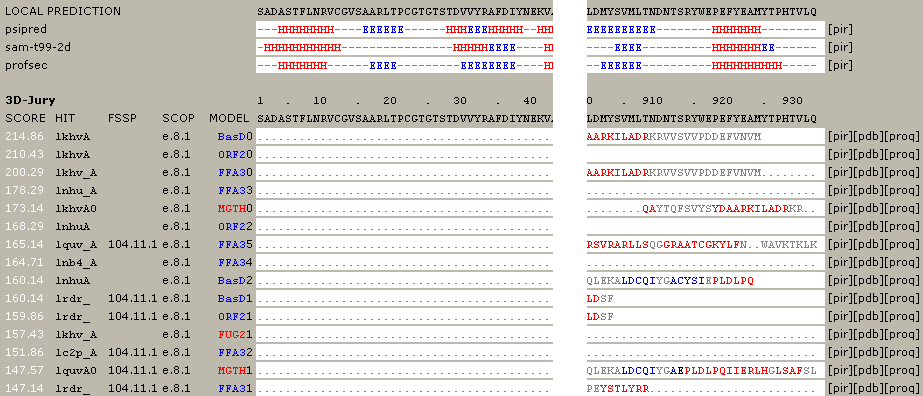
3. The results from different servers were scored by their "3D-Jury" system. In the default setting, the top predicator (well, according the the 3D-Jury score) is BasD. Several top scorers have identified 1khv as the template structure for nsp9. The PDB code 1khv corresponds to crystal structure of RNA-dependent RNA polymerase of a RNA virus, Rabbit hemorrhagic disease virus (RHDV). Also notice that only the C-terminal region of nsp9 can be aligned with 1khv, suggesting the ~500 residues at the C-terminal of nsp9 may be a polymerase domain.
4. You can download the CA trace by clicking the [pdb] button on the right. If you have the license key for MODELLER, you can ask the server to build the full-atom model by clicking the [prog] button. You can obtained the license key for MODELLER at http://salilab.org/modeller/. Academic license is free of charge.
II. Model Building -
1st trial
1. In this tutorial, we will stick to SWISS-MODEL for model building. 2. Firstly, the N- and C-terminal residues were trimmed to simplify the alignment:

4. Now start SWISS-PDBViewer by double-clicking spdbv.exe. Load the primary sequence (nsp9C.fas) and the template structure (1khvA.pdb) to the program. I have converted the above alignment in foldfit format. After loading the foldfit alignment, you will notice the structure of nsp9 is threaded onto the structure of 1khvA.
5. Now submit the modelling request to SWISS-MODEL.
III. Model evaluation
1. Until now, we are able to predict structure with only minimal human intervention. It may give you a false impression that protein structure can be predicted automatically. However, never trust the automatically generated structure - you should inspect the alignment and the predicted structure manually to avoid obvious mistakes and to improve the prediction.2. To save time, I have done the 1st round model building and the result can be found here (round1.pdb).
3. Now check the alignment. In particular, pay attention to gaps in the alignment. There is a big gap between Ser-335 and Asp-354 (residue numbers correspond to the sequence of 1khv) . This results in insertion of a big loop between strand A and B (see figure):
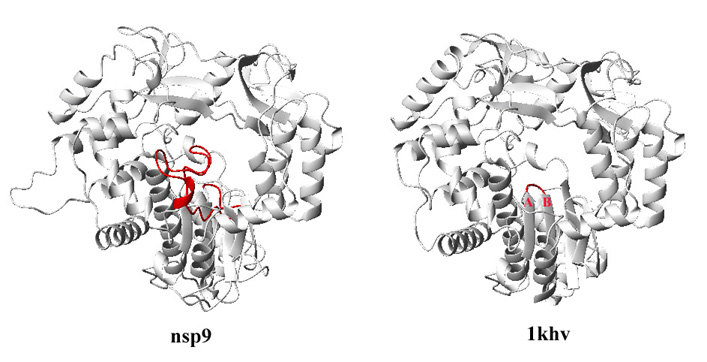
4. Is this insertion reasonable? How do you judge whether it is reasonable or not? It is time to do some literature search. More you understand the target, better the chance you predict the structure correctly. In fact, Asp-354 and Asp-355 are the active site residues of the polymerase. The insertion, which will block the active site, is very likely to be wrong.
5. Now it is time to check the result from bioinfo.pl again. Here is the alignment of the region for the top 10 scorers of bioinfo.pl server:

In this region, the alignment of the 3rd scorer is better. In this case, the insertion are moved away from the active site:

6. You can resubmit the job the SWISS-MODEL to see your result. The result is shown here.